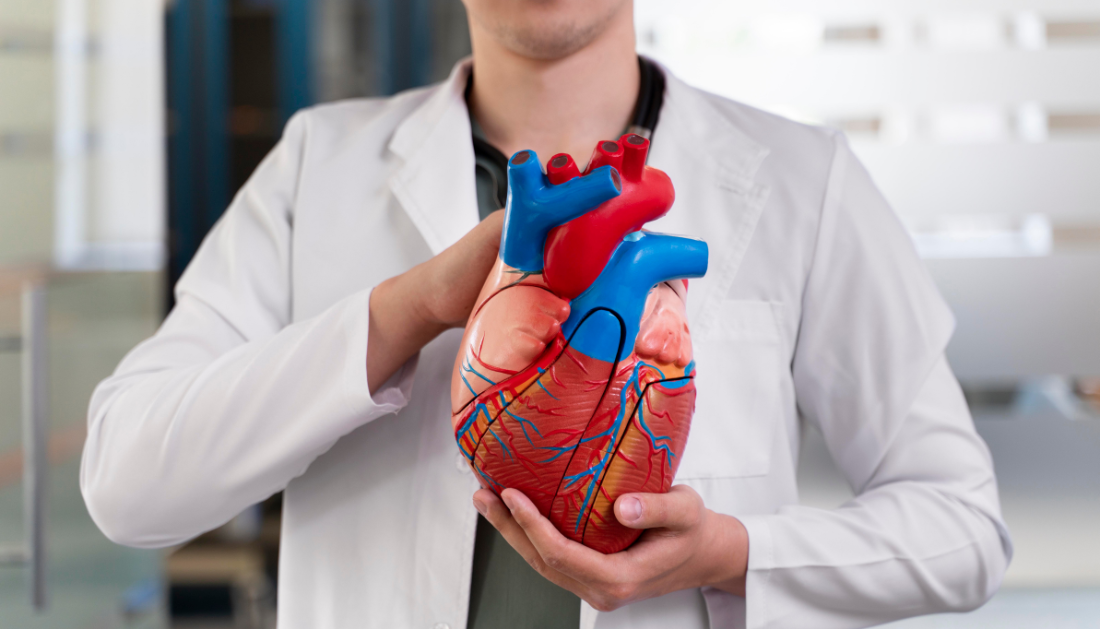
Study: m6A reader YTHDF1 promotes cardiac fibrosis by enhancing AXL translation
In cardiac fibrosis, a disorder marked by an excessive buildup of extracellular matrix proteins that causes the heart to harden and lose function, the function of the m6A reader YTHDF1 is examined in this article. A common pathogenic process in a number of cardiovascular disorders, such as heart failure, myocardial infarction, and hypertension, is cardiac fibrosis.
The protein known to bind to N6-methyladenosine (m6A), a common internal alteration in eukaryotic messenger RNA (mRNA), is the subject of the investigation. Numerous biological activities, such as mRNA splicing, transport, translation, and degradation, have been linked to m6A alteration. According to the study, YTHDF1 increases the translation of AXL, a receptor tyrosine kinase linked to tissue fibrosis and healing, which in turn causes cardiac fibrosis.
Studies carried out during the investigation show that YTHDF1 interacts with AXL mRNA, resulting in an elevation in AXL protein levels. It has been demonstrated that this connection is essential for the fibrotic response in cardiac fibroblasts, which are the cells that make up the extracellular matrix in the heart. Additionally, the study discovers that YTHDF1 overexpression
The m6A modification status of AXL mRNA controls YTHDF1-mediated AXL translation, according to additional research into the underlying molecular mechanisms. According to the study, YTHDF1 binds to the m6A-modified AXL mRNA to aid in its translation into protein, which in turn stimulates the activation of fibroblasts and the synthesis of collagen.
The involvement of YTHDF1 in cardiac fibrosis is supported by in vivo research employing animal models. These studies also demonstrate that YTHDF1 inhibition or deletion reduces cardiac fibrosis, indicating YTHDF1 may be a viable target for therapy. The results underline the significance of the m6A RNA methylation pathway in cardiac fibrosis and suggest that YTHDF1 targeting may represent a unique therapeutic approach for this condition.
The study also addresses the wider ramifications of these results for other illnesses such kidney, lung, and liver cirrhosis where fibrosis is a major factor. Comprehending the function of m6A modification and its receptors, such as YTHDF1, in these mechanisms may facilitate the creation of novel therapeutic strategies for various fibrotic disorders.
To sum up, the research offers valuable understanding of the molecular processes that underlie this condition and identifies YTHDF1 as a crucial modulator of the condition. YTHDF1 stimulates the activation of cardiac fibroblasts and the consequent development of fibrosis by improving AXL translation. These results contribute to our understanding of the pathophysiology of cardiac fibrosis and open up new possibilities for the development of tailored therapeutics for the treatment of this and probably other fibrotic conditions.
For more information: m6A reader YTHDF1 promotes cardiac fibrosis by enhancing AXL translation, Frontiers of Medicine. doi.org/10.1007/s11684-023-1052-4
 Macrophages Attack Live Melanoma Cells in Real Time
Macrophages Attack Live Melanoma Cells in Real TimeKey Takeaways Researchers at the Garvan.
Diseño y navegación pensados para la.
 Facial Micromovements Linked to Accurate Pain Detection
Facial Micromovements Linked to Accurate Pain DetectionKey Takeaways Researchers at Rutgers University–New.
 Nicotine Pouches Under Scrutiny in New WHO Warning
Nicotine Pouches Under Scrutiny in New WHO WarningKey Takeaways The World Health Organization.
 Cannabis and Tobacco Co-Use May Increase Psychosis Risk
Cannabis and Tobacco Co-Use May Increase Psychosis RiskKey Takeaways A new study in.
 Skin Cancer Risk After Transplant Needs Better Screening
Skin Cancer Risk After Transplant Needs Better ScreeningKey Points Summary Solid organ transplant.
 Polygenic Risk Scores Improve Early Heart Disease Detection
Polygenic Risk Scores Improve Early Heart Disease DetectionKey Points A new multicondition polygenic.
 Prenatal Sedatives Psychiatric Risk: New Study Findings
Prenatal Sedatives Psychiatric Risk: New Study FindingsQuick Summary A large South Korean.
 Cardiovascular Health from Early Life Lowers Disease Risk
Cardiovascular Health from Early Life Lowers Disease RiskKey Points Summary: Cumulative cardiovascular health.
 Early Autism Detection Using Wearable Sensors in Infants
Early Autism Detection Using Wearable Sensors in InfantsKey Highlights: Wearable movement sensors may.
Leave a Comment