Image by brgfx on Freepik
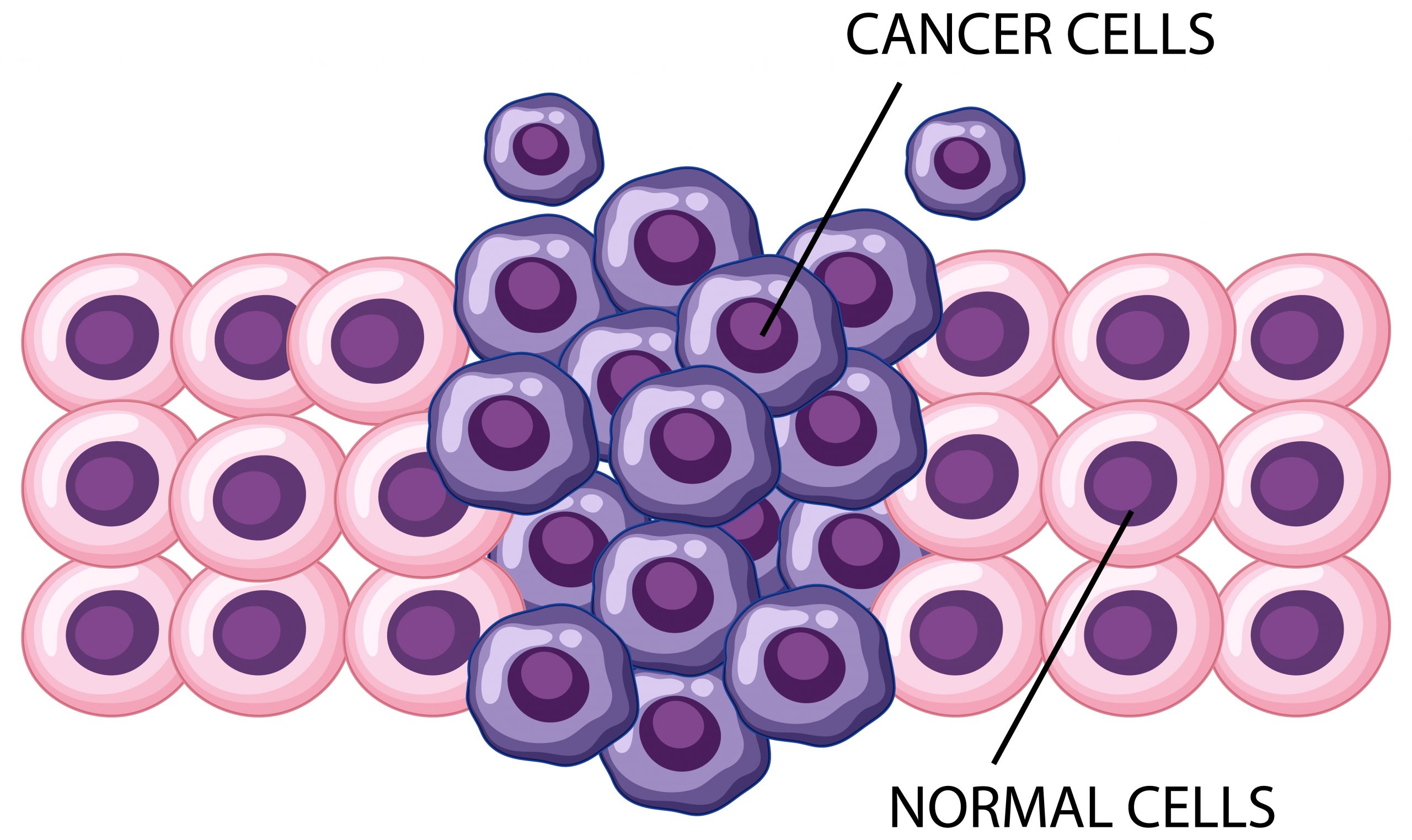
The loss of a gene called SYNCRIP in prostate cancer tumors activates cellular machinery that causes random mutations throughout the genome, leading to resistance to targeted treatments, according to a team led by UT Southwestern Medical Center researchers. The results, which were published in Cancer Cell, could lead to new therapies that disrupt this process in prostate and other cancer types, making them significantly more treatable.
“This study paves the way for innovative strategies to fix the broken ‘brake’ for mutagenesis in cancer and curb resistance to treatments,” said study leader Ping Mu, Ph.D., Assistant Professor of Molecular Biology at UT Southwestern and a member of the Harold C. Simmons Comprehensive Cancer Center.
Researchers have long known that many cancers are caused by DNA abnormalities, and that more mutations regularly arise, causing tumors to grow and spread. Although medications exist to target tumors with specific mutations, they do not address the entire spectrum of alterations that occur in cancer cells, according to Dr. Mu. As a result, the more mutations a tumor has, the more resistant it is to treatment. However, it is unknown how and why these extra mutations occur so quickly.
Dr. Mu and his colleagues sought genetic differences in the cells of prostate cancer tumors – the Mu lab’s focus – from samples taken from xenografted human tumors before and after antiandrogen therapy, a common treatment that deprives prostate cancers of sex hormones that can fuel their growth. Although this treatment is frequently effective at first, prostate cancers almost always develop resistance to antiandrogen therapy with time.
The SYNCRIP gene had vanished in almost half of the post-treatment samples. The researchers also discovered a number of mutations that bore the signature of APOBEC proteins, which are pieces of cellular machinery that produce a specific form of mutation used for immunological protection in healthy cells.
Further research suggests that SYNCRIP acts as a brake for APOBEC3B, a prominent member of the APOBEC protein family, keeping its activity under check. Dr. Mu noted that when SYNCRIP is lost, APOBEC3B’s function becomes unregulated, resulting in random mutations throughout the genome. Even in the absence of SYNCRIP, cells genetically engineered to eliminate APOBEC3B did not develop mutations, suggesting that their roles are connected.
The researchers analyzed the genomes of post-treatment cells for APOBEC-induced mutations that caused genes to become hyperactive in search of genetic drivers of treatment resistance that originate from this process. They discovered eight genes that appear to be hotspots for APOBEC-driven mutations, and all of them rendered antiandrogen therapy ineffective. The cells became sensitive to therapy again when the researchers removed these genes from them.
Because APOBEC hallmark mutations are prevalent in over 70% of human tumors, the findings shed light on various oncology puzzles, according to Dr. Mu. These include why carcinogenic tumors tend to develop so many mutations so quickly and why the mutation pattern varies between people with the same cancer type and, in some cases, between cells in the same tumor.
Currently, tumors are treated by treating these mutations individually, which is successful only if a targeted treatment for a specific mutation exists, he added. Researchers may be able to design medicines that restore SYNCRIP or suppress APOBEC proteins in the future to prevent resistance-driving mutations from developing.
 Prenatal Smoking Raises Risk of Child Mental Disorders
Prenatal Smoking Raises Risk of Child Mental DisordersKey Summary Prenatal smoking is associated.
 Physical Activity Guidelines Gap: Walking is Insufficient
Physical Activity Guidelines Gap: Walking is InsufficientQuick Summary Walking remains the most.
 Breast Cancer Risk Rises with Aging Tissue Changes
Breast Cancer Risk Rises with Aging Tissue ChangesKey Highlights A 3-million-cell atlas reveals.
 Type 2 Diabetes Risk Rising in Genetically Susceptible
Type 2 Diabetes Risk Rising in Genetically SusceptibleQuick Summary Rising type 2 diabetes.
 Female Microbiome Shaped by Diet, Stress, Unhealthy Lifestyle
Female Microbiome Shaped by Diet, Stress, Unhealthy LifestyleQuick Summary Lifestyle factors significantly influence.
 Transcatheter Valve-in-Valve Improves Mitral Outcomes
Transcatheter Valve-in-Valve Improves Mitral OutcomesKey Highlights Transcatheter mitral valve-in-valve (mVIV).
 Stroke Rehabilitation: Early High-Intensity Therapy Findings
Stroke Rehabilitation: Early High-Intensity Therapy FindingsKey Highlights High-intensity therapy within 2.
 TRPM8 Cold Sensation Mechanism Explained for Pain Care
TRPM8 Cold Sensation Mechanism Explained for Pain CareQuick Summary TRPM8 ion channel converts.
 Gum Recession from Snus Confirmed, Caries Risk Debated
Gum Recession from Snus Confirmed, Caries Risk DebatedKey Highlights Snus use is strongly.
 Hypertensive Disorders of Pregnancy: Role of Daily Activity
Hypertensive Disorders of Pregnancy: Role of Daily ActivityKey Points Summary Limiting sedentary time.
Leave a Comment